Ladda ner presentationen
Presentation laddar. Vänta.

1
Myndigheterna och läkemedelsindustrin ett mycket reglerat samarbete Marianne Andersson and Charlotta Klockare Regulatory Affairs, AstraZeneca R&D Mölndal

2
FÖRKORTNINGAR CMCChemistry, Manufacturing and Controls CMSConcerned Member State CPCentralised Procedure CTAClinical Trial Application CTDCommon Technical Document DCPDecentralised Procedure EMEA European Agency for the Evaluation of Medicinal Products EFPIAEuropean Federation of Pharmaceutical Industries Association FDAFood and Drug Administration (USA) GCPGood Clinical Practice GLPGood Laboratory Practice GMPGood Manufacturing Practice ICHInternational Conference on Harmonisation LIFLäkemedelsindustriföreningen MAAMarketing Authorisation Application MAHMarketing Authorisation Holder MHLWMinistry of Health, Labour and Welfare (Japan) MPAMedical Products Agency (Sweden) MRPMutual Recognition Procedure NDANew Drug Application PSURPeriodic Safety Update Report R&DResearch & Development SmPCSummary of Product Characteristics WHOWorld Health Organisation
 GCPGood Clinical Practice GLPGood Laboratory Practice GMPGood Manufacturing Practice ICHInternational Conference on Harmonisation LIFLäkemedelsindustriföreningen MAAMarketing Authorisation Application MAHMarketing Authorisation Holder MHLWMinistry of Health, Labour and Welfare (Japan) MPAMedical Products Agency (Sweden) MRPMutual Recognition Procedure NDANew Drug Application PSURPeriodic Safety Update Report R&DResearch & Development SmPCSummary of Product Characteristics WHOWorld Health Organisation")
3
International Conference on Harmonisation ICH Ett nätverkt - Ingen myndighet. Myndigheterna inom EU, USA och Japan deltar tillsammans med organisationer som företräder läkemedelsindustrin inom varje region samt Akademin. Enas om riktlinjer som senare implementeras i de regionala lagarna och reglerna “Quality, Safety, Efficacy”, GMP, GCP, GLP WHO är ej medlem utan deltar bara som observatör. Andra länders myndigheter följer ICH arbetet. T.ex. Kanada och Australien

4
ICH – Exempel på Riktlinjer Stabilitet - produkt och i förpackning Föroreningar – vid tillverkning, i produkt Specifikationer – av substans, produkt Good Manufacturing Practices (GMP) Good Clinical Practices (GCP) Good Laboratory Practices (GLP) Farmaceutisk utvecklingsarbete Riskhantering – system, planer, rapportering Common Technical Document (CTD) - struktur
 Good Clinical Practices (GCP) Good Laboratory Practices (GLP) Farmaceutisk utvecklingsarbete Riskhantering – system, planer, rapportering Common Technical Document (CTD) - struktur")
5
Vilka är läkemedelsmyndigheterna De vi interagerar mest med från R&D: EU EMA (European Medicines Agency) Nationella myndigheter t.ex. Läkemedelsverket (LV el. MPA) USA FDA (Food and Drug Administration) Japan MHLW (Ministry of Health, Labour & Welfare) Kina SFDA (State Food and Drug Administration) and CDE (Drug Evaluation Center) Men det finns ju många fler, bl.a.: Australien TGA (Therapetic Goods Administration) Ryssland Federal Service on Surveillance in Healthcare and Social Development
 USA FDA (Food and Drug Administration) Japan MHLW (Ministry of Health, Labour & Welfare) Kina SFDA (State Food and Drug Administration) and CDE (Drug Evaluation Center) Men det finns ju många fler, bl.a.: Australien TGA (Therapetic Goods Administration) Ryssland Federal Service on Surveillance in Healthcare and Social Development.")
6
Läkemedelsmyndigheternas roll Utvärdera ansökningar (“Market Authorisation Application”,MAA el NDA) för nya läkemedel Har läkemedlet tillfredställande Effekt? Är Säkerheten tillfredställande? Fördelaktig nytta/risk balans? Är “Quality” (Kemi, tillverkning och kontroll av produkt) tillfredställande? Utfärda godkännande för marknadsföring av ett läkemedel (eller avslå ansökan) Granska och godkänna/avslå utförandet av kliniska studier Säkerställa att förordningarna och lagstiftning följs Övervaka/inspektera tillverkning Utveckla läkemedelslagstiftningen
 tillfredställande. Utfärda godkännande för marknadsföring av ett läkemedel (eller avslå ansökan) Granska och godkänna/avslå utförandet av kliniska studier Säkerställa att förordningarna och lagstiftning följs Övervaka/inspektera tillverkning Utveckla läkemedelslagstiftningen.")
7
‘Registreringsavdelningens’ roll inom industrin Tolkning av regelverk och råd till projektgrupper: Vilka krav finns, vad måste göras, hur ska det göras och när? Kontaktyta mot myndigheter Utveckla ”märkningen” Vad kan vi påstå, vad måste vi säga Kliniska prövningsansökningar Ansöka om vetenskaplig rådgivning Registreringsansökningar – nya läkemedel Svara på frågor, utreda krav, granska dokumentation Sköta licensunderhåll och utveckla Omregistrering, safety rapportering, ansöka om ändringar

8
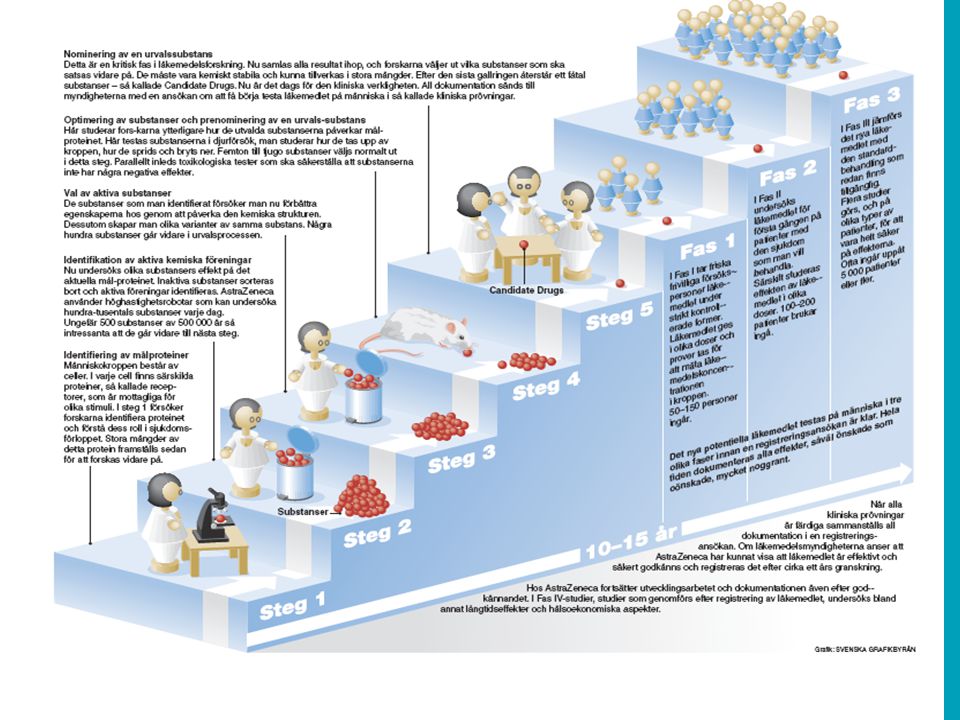
Ett läkemedels livscykel Utveckling Licens Underhåll Fas I II III Vetenskaplig rådgivning MAA Produkt-resumé Säkerhetsövervakning Produktutveckling Nya indikationer, nya formuleringar ’Candidate Drug’ val Prekliniska försök/toxstudier Formulering för kliniska studier Första studier i människa Dos-val, konceptstudier Fas IV Stora Effektstudier Formulering för marknadsberedning Uppskalning av tillverkning

9
Vetenskaplig rådgivning Begränsat till vetenskapliga frågeställningar Ska ange de specifika frågorna och bakgrund till dessa Råd från myndighet är inte tvingande men man måste kunna förklara/motivera om man ej följer Myndigheten kan ge muntliga eller skriftliga råd

10
Det kliniska utvecklingsprogrammet Fas I (Human Pharmacology) >10-tals friska frivilliga – säkerhet, farmakokinetik och -dynamik. Fas II (Therapeutic exploratory) >100 talet patienter – testa konceptet och dos- intervall. Fåtal länder och kliniker Fas III (Therapeutic confirmatory) >1000 tals patienter – stora studier, många länder, kliniker. Outcome studier. Fas IV (Therapeutic use) 10000-tals patienter – ”post marketing studies”
 >100 talet patienter – testa konceptet och dos- intervall. Fåtal länder och kliniker Fas III (Therapeutic confirmatory) >1000 tals patienter – stora studier, många länder, kliniker. Outcome studier. Fas IV (Therapeutic use) tals patienter – post marketing studies .")
11
Klinisk prövning på patienter Studieprotokollet Syftet, vad ska göras, på vilka, hur många och hur ska allt göras? Val av länder, kliniker och patienter Ansökan om tillstånd Monitorering, följsamhet till protokoll Kontroll av data Avblindning och statistisk analys Rapportering av resultat

12
Hur ser en registreringsansökan ut? Krav på Format/Struktur = Common Technical Document (CTD) Elektronisk CTD Krav på Innehåll = Ansökningsformulär Betalda avgifter Dokumentationen
 Elektronisk CTD Krav på Innehåll = Ansökningsformulär Betalda avgifter Dokumentationen.")
13
CTD strukturen (formatet) Module 1 Regional Administrative Information 1.0 CTD ToC* 2.1 CTD Introduction 2.2 Quality Overall Summary 2.3 Nonclinical Overview 2.4 Nonclinical Summary 2.6 Clinical Overview 2.5 Clinical Summary 2.7 Module 3 Quality Module 4 Nonclinical Study reports Module 5 Clinical Study reports Module 2 Not part of CTD CTD *ToC = Table of Contents
 Module 1 Regional Administrative Information 1.0 CTD ToC* 2.1 CTD Introduction 2.2 Quality Overall Summary 2.3 Nonclinical Overview 2.4 Nonclinical Summary 2.6 Clinical Overview 2.5 Clinical Summary 2.7 Module 3 Quality Module 4 Nonclinical Study reports Module 5 Clinical Study reports Module 2 Not part of CTD CTD *ToC = Table of Contents")
14
Ansökan om licens och Procedurer inom EU USA: New Drug Application (NDA) Japan: Japanese New Drug Application (JNDA) Europa: Marketing Authorisation Application (MAA) 3 olika procedurer Centralised Procedure Decentralised Procedure National Procedure
 Japan: Japanese New Drug Application (JNDA) Europa: Marketing Authorisation Application (MAA) 3 olika procedurer Centralised Procedure Decentralised Procedure National Procedure")
15
RISK BENEFIT Quality Efficacy Safety Utvärdering av läkemedel

16
Produktinformationen Basen för marknadsföring Produktresumén (Summary of Product Characteristics, SmPC) Information till förskrivare Liknar FASS-text (annan disposition) Bipacksedeln och märkning Information till användare/patienten För exempel se Läkemedelsverkets hemsida, ”sök läkemedelsfakta” eller fass.se Förhandlingar under granskningsperioden
 Information till förskrivare Liknar FASS-text (annan disposition) Bipacksedeln och märkning Information till användare/patienten För exempel se Läkemedelsverkets hemsida, sök läkemedelsfakta eller fass.se Förhandlingar under granskningsperioden")
17
Ansökningar efter första godkännande Ändringar (Variations): Typ IA, IB och Typ II – olika krav: t.ex. Namnbyte, adressbyte av packsite (Typ IA) Nytt site för tillverkning, packning (Typ IB) Nya tillverkningsmetoder (Typ II) Ändring av produktinformation (Typ II) Produktutveckling Ny formlering eller administreringssätt Ny styrka Ny patientpopulation, indikation
 Nytt site för tillverkning, packning (Typ IB) Nya tillverkningsmetoder (Typ II) Ändring av produktinformation (Typ II) Produktutveckling Ny formlering eller administreringssätt Ny styrka Ny patientpopulation, indikation.")
18
Säkerhetsdokumentation och övervakning ett nyckelområde ”Pharmacovigilance”: Kontinuerlig insamling och rapportering av biverkningar, “PSUR” Monitorera spontana rapporter, kliniska studier, litteraturen, internet. Kontinuerlig nytta/risk utvärdering Om och när så krävs, vidta åtgärder och informera myndigheter och läkare Urgent Safety Restrictions Lagstiftning, regelverk!

Liknande presentationer




 roll>")














 ansvaret för tillsyn av hälso- och sjukvård, socialtjänst och.>")