Ladda ner presentationen
Presentation laddar. Vänta.

1
Benign hematologi Åsa Derolf Hematologiskt Centrum Karolinska Universitetssjukhuset

2
Vad betyder benign hematologi? Anemier utan bakomliggande malign blodsjukdom – Hemolys – Hemoglobinopatier – PNH – enzymdefekter Immunologisk trombocytopen purpura (ITP)
.")
3
Hemolys För tidig destruktion av erytrocyter Kompensering (EPO) - Erythroid hyperplasi i BM Retikulocytos Anemi när benmärgen inte kan kompensera med nybildning Akut, Kronisk eller episodisk hemolys Vissa har kronisk hemolys som är kompenserad, men vid tex infektion eller stress utvecklas anemi
 - Erythroid hyperplasi i BM Retikulocytos Anemi när benmärgen inte kan kompensera med nybildning Akut, Kronisk eller episodisk hemolys Vissa har kronisk hemolys som är kompenserad, men vid tex infektion eller stress utvecklas anemi")
4
Hemolytisk anemi Hemolys är vanligen extravasal dvs makrofager i mjälte, benmärg och lever fagocyterar erytrocyter Ovanligt är intravasal hemolys då man får hemoglobinuri. HEMOGLOBINURI fritt hemoglobin i urinen pga lyserade trasiga röda blodkroppar som läcker. HEMATURI röda blodkropparna är intakta i urinen. Båda dessa ger mörk urin.

5
När misstänka hemolys? anemi högt LD Högt bilirubin Ökad mängd retikulocyter Omätbart haptoglobin

6
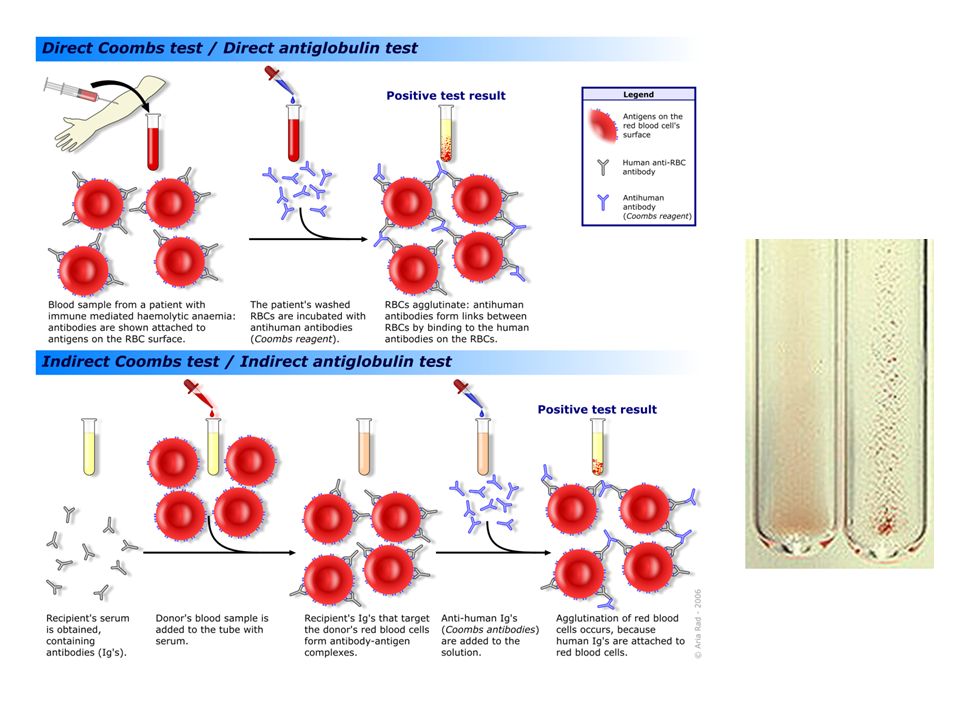
Autoimmun hemolys Antikroppar mot erytrocyter (antigen på cellmembran) Leder till destruktion (mjälte) Hemolysprover positiva Autoantikroppar påvisas via Coombsprov / DAT
 Leder till destruktion (mjälte) Hemolysprover positiva Autoantikroppar påvisas via Coombsprov / DAT")
8
Immunohemolys Varma antikroppar IgG (IgA) Aktiva vid 37°C Ofta idiopatiskt. Hematologiskt malignitet? Övriga autoimmunsjukdomar - SLE Opsonisation – phagocytos Svarar ofta på beh med kortison Ev splenektomi Kalla antikroppar IgM (low affinity) Binds vid ≤ 32°C Fingrar, tåar Komplementbindning – IgM lossnar – C3b kvar (opsonin) – RES – hemolys Ofta mild sjukdom Raynauds phenomenon Svarar dåligt på kortison Antikroppsbeh / cytostatika
 Binds vid ≤ 32°C Fingrar, tåar Komplementbindning – IgM lossnar – C3b kvar (opsonin) – RES – hemolys Ofta mild sjukdom Raynauds phenomenon Svarar dåligt på kortison Antikroppsbeh / cytostatika.")
9
DAT-negativ hemolys Hereditär sfärocytos Hemoglobinopatier PNH (paroxysmal nocturnal hemoglobinuri) TTP (trombotisk trombocytopen purpura) Läkemedelsutlöst Enzymdefekter – G6PD-brist (favism) – pyruvatkinasbrist
 TTP (trombotisk trombocytopen purpura) Läkemedelsutlöst Enzymdefekter – G6PD-brist (favism) – pyruvatkinasbrist")
10
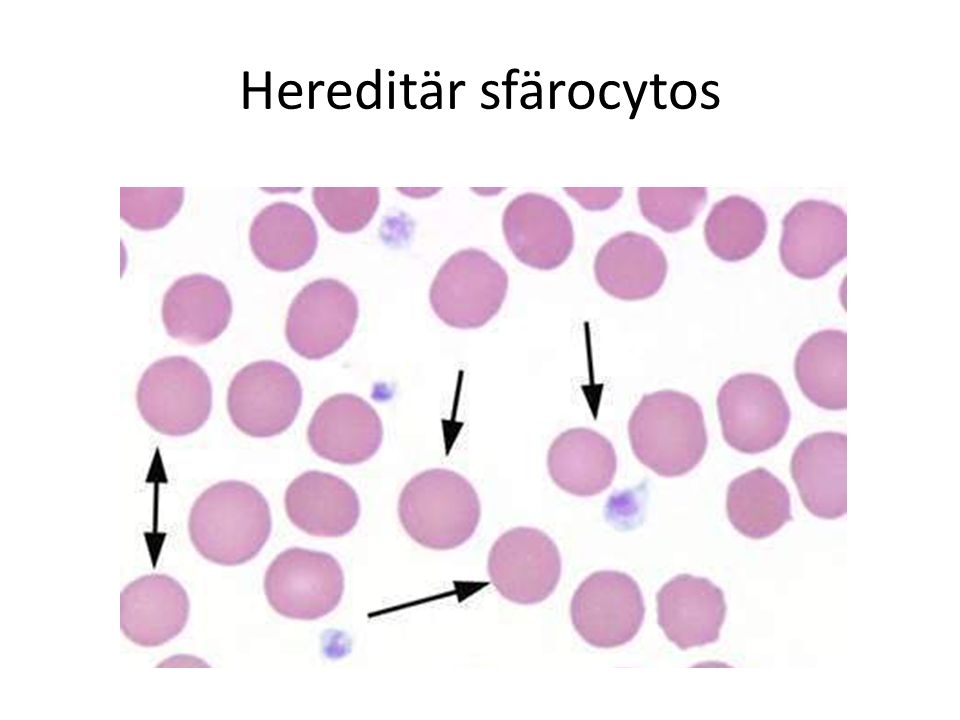
Hereditär sfärocytos Defekt i cellskelettet Erytrocyterna – Tåler dåligt att deformeras – Förlurar cellmembran gradvis – Blir bollformade (sfärocyter) – Tas upp i mjälten Anemi – splenomegali – ikterus – gallstenar Splenektomi botar ofta anemin Nedsatt osmolar resistans / ökad fragilitet
 – Tas upp i mjälten Anemi – splenomegali – ikterus – gallstenar Splenektomi botar ofta anemin Nedsatt osmolar resistans / ökad fragilitet")
11
Struktur Cellmembran Cellskelett Spectrin Actin Protein 4.1 Ankyrin

12
Hereditär sfärocytos

14
Hemoglobin Hemoglobinmolekylen hos vuxna är uppbyggd av fyra globinkedjor, två alfa- (α) och två betakedjor (β).
 och två betakedjor (β).")
15
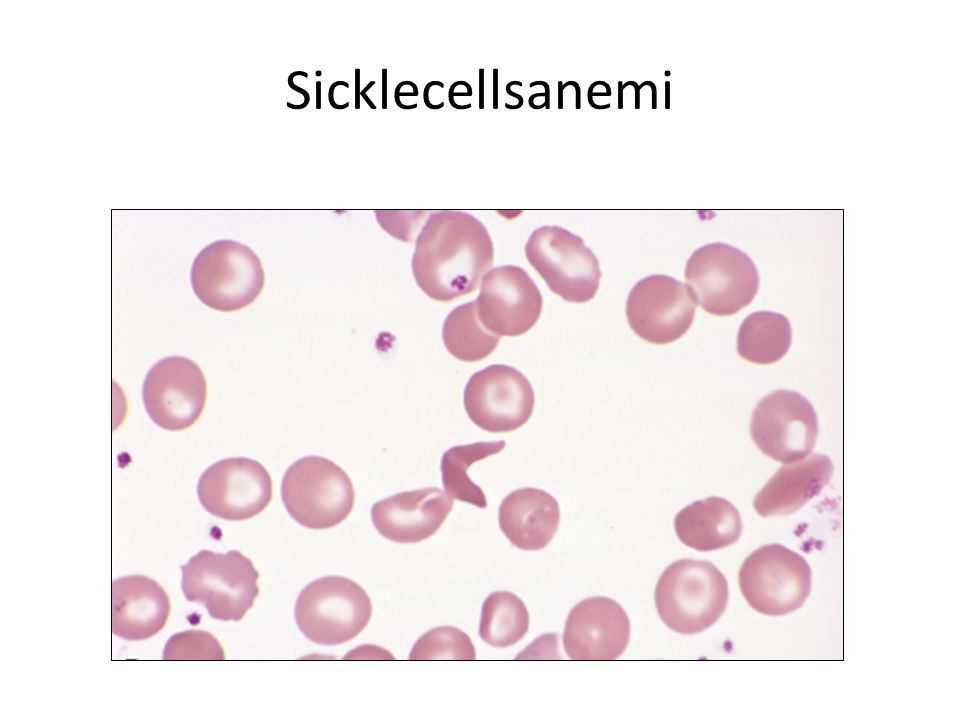
Sicklecellsanemi Defekt i β-globin-kedja HbS – autosomal recessive mutation – Mer vanligt i malariaområden HbS “klibbar ihop sig” med varandra när släpper syret = gelation, crystallization Formförändring i erytrocyter (sickling) HbSS /HbSC
 HbSS /HbSC")
16
Sicklecellsanemi

18
Formförändringen reversibel (syrebindning) Men erytrocyterna blir snabbt trasiga – Skada i cellmembran – Dehydrerade – Styva och oformbara – Tas upp i RES – Förkortad livslängs (20 d). Kan leda till anemi Kan orsaka stas i mikrocirkulation ischemi

19
Sicklecellsanemi Mjälte – Först förstoring – Sedan ischemi – Fibros – Funktionell hyposplenism - ökad infektionsrisk

20
Sicklecellsanemi Hos homocygota visar sjukdomen sig omkring 6 mån ålder Därefter kronisk hemolys Anemi och vävnadsskada Försämringar – “kriser” F f a smärtkriser – Ofta p g a försämrad cirkulation till BM

21
Sicklecellsanemi

22
Sickle-cellskris Utlöst av kyla/infektion/dehydrering/hypoxi Muskel- och skelettsmärtor – kräver höga doser opioider Vätska Syrgas Blodtransfusion för att uppnå patientens normala nivå Behandla ev infektion, frikostigt med antibiotika

23
Acute chest syndrome Vanligaste dödsorsaken bland patienter med sickle-cellanemi Bröstsmärta, lunginfiltrat Orsaker till stora delar okänd Symtomatisk behandling – hög vårdnivå

24
Behandlingar Hydrea – ökar mängden HbF Erytraferes – minskar andelen HbS Regelbundna transfusioner Stamcellstransplantation Kelerande behandling

25
Talassemi Vid talassemi bildas för litet av endera α- kedja (α-talassemi) eller β-kedja (β-talassemi). Den kedja som finns i överskott bildar tetramerer som stör tidig erytropoes i benmärgen (ineffektiv erytropoes) och/eller bidrar till att erytrocyterna elimineras i mjälten (hemolys).) α-thalassemia – Minnskad produktion av α-kedjor β-thalassemia – Minskad produktion av β –kedjor (β + ) – Ingen produktion av β-kedjor (β 0 )
 och/eller bidrar till att erytrocyterna elimineras i mjälten (hemolys).) α-thalassemia – Minnskad produktion av α-kedjor β-thalassemia – Minskad produktion av β –kedjor (β + ) – Ingen produktion av β-kedjor (β 0 ).")
26
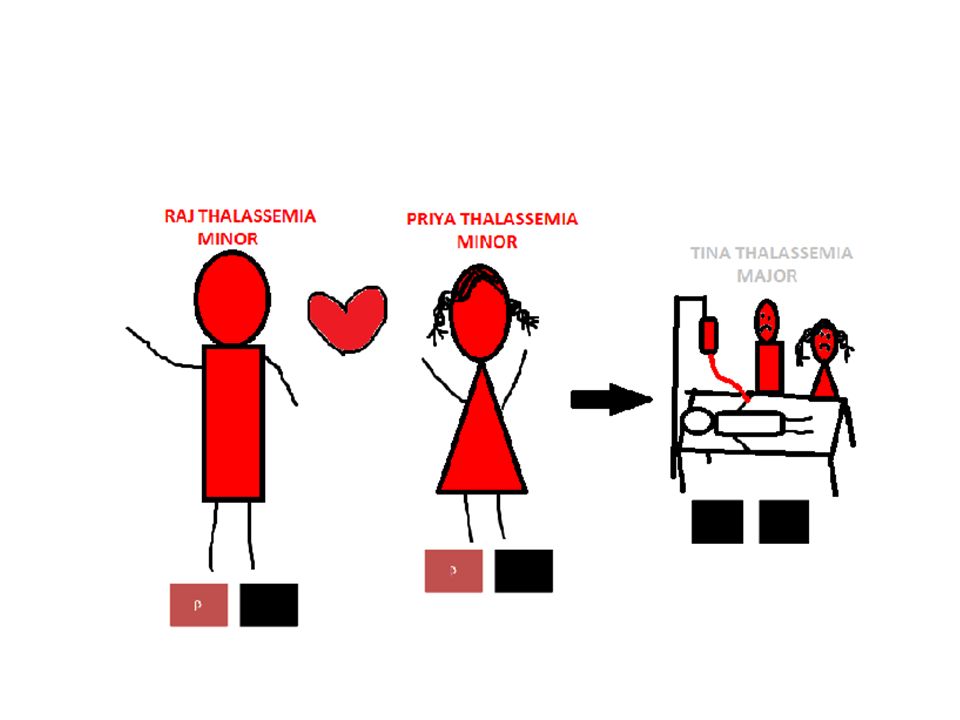
β-talassemi Genen för bildning av β-kedjor sitter på kromosom 11. Mer än 200 olika mutationer kan stoppa bildningen av β-kedjor helt (β0), reducera till 10 % (β+) eller med olika % -satser (β++). Ärvs en β0-allel från vardera föräldern uppstår β- talassemia major med helt utslagen bildning av β- kedjor (β0β0). Kombinationer med kraftigt nedsatt bildning av β- kedjor (β0β+ eller β+β+) resulterar i β-talassemia intermedia ( β -TI) Vid minst 50 % bevarad bildning av β-kedjor ger kliniskt indolent bärarskap (β0 β++) sk talassemia minor.
, reducera till 10 % (β+) eller med olika % -satser (β++). Ärvs en β0-allel från vardera föräldern uppstår β- talassemia major med helt utslagen bildning av β- kedjor (β0β0). Kombinationer med kraftigt nedsatt bildning av β- kedjor (β0β+ eller β+β+) resulterar i β-talassemia intermedia ( β -TI) Vid minst 50 % bevarad bildning av β-kedjor ger kliniskt indolent bärarskap (β0 β++) sk talassemia minor..")
28
β-Talassemi Konsekvens (homocygot) – Minskad HbA produktion – Mikrocytäre ( MCV) och hypokroma ( MCHC) erytrocyter – Hemolys. α-kedjor klibbar ihop sig intracellulärt. Onormala erytrocyter som tas upp i RES – Erytroblaster dör i BM (intramedullärt) Inefektiv erytropoes – Skelettdeformationer
 Inefektiv erytropoes – Skelettdeformationer.")
29
β-Talassemi

30
α –Talassemi Oftast deletion i α-globin genen 4 möjligheter: – -α/αα: Bärare – -α/-α: Trait (thalassemia minor) – --/-α: HbH (β 4 ) tetramer (haemolys) – --/--: hydrops fetalis Kliniken liknar β-Talassemi men oftast mildare
 – --/-α: HbH (β 4 ) tetramer (haemolys) – --/--: hydrops fetalis Kliniken liknar β-Talassemi men oftast mildare")
31
Andra hemoglobinvarianter HbAE och HbEE kan kliniskt te sig som en β-talassemia minor. HbE/β0kan te sig som en talassemia major och HbS/β0 som en sickelcellssjukdom. Också α och β nedärvs oberoende av varandra så man kan få ”hybriden” αβ i olika former.

32
Talassemi

33
Mekaniskt hemolys Många olika orsaker – Mikroangiopatier T ex DIC (fibrintrådar) – Makrovaskulär: oftast mekanisk hjärtklaff Behandla underliggande orsak TTP – brist på ADAMTS13 – multimerer av vWf klyver erytocyter – Hemolys, trombocytopeni, njurpåverkan, medvetandepåverkan
 – Makrovaskulär: oftast mekanisk hjärtklaff Behandla underliggande orsak TTP – brist på ADAMTS13 – multimerer av vWf klyver erytocyter – Hemolys, trombocytopeni, njurpåverkan, medvetandepåverkan")
34
Mekanisk hemolys

35
PNH Incidens 1,3/ 1 miljon invånare De vanligaste kliniska manifestationerna vid klassisk PNH är hemolytisk anemi, tromboseroch benmärgssvikt. Diagnos – Hemolysbild – flödescytoemtri på blod CD55 och CD59 – Benmärgsprov för att utesluta annan blodsjukdom

36
Komplementhämmare på normala röda blodkroppar skyddar cellen mot komplementattacker. Vid PNH har man en mutation ( PIGA genen på X kromosomen) så man inte kan göra ett GPI ankare, då kan inte kpmplementhämmarna fästa på röda blodkroppen. Därför kan komplement lysera röda blodkropparna. Detta sker direkt i blodbanan, intravasal hemolys. Detta sker extra mycket i sur miljö, tex vid hypoventilation (hög höjdsvistelse, nattetid). PNH -Intravasal hemolys
 så man inte kan göra ett GPI ankare, då kan inte kpmplementhämmarna fästa på röda blodkroppen. Därför kan komplement lysera röda blodkropparna. Detta sker direkt i blodbanan, intravasal hemolys. Detta sker extra mycket i sur miljö, tex vid hypoventilation (hög höjdsvistelse, nattetid). PNH -Intravasal hemolys.")
37
PNH Intravasal lys av erytrocyterna ger fritt hemoglobin i plasma vilket binds irreversibelt till kväveoxid (NO). Det låga NO:t ger ökad tonus i glatt muskulatur med vasokonstriktion vilket leder till sammandragning i tarmen (buksmärtor), pulmonell hypertension, erektil dysfunktion, spasm i esofagus (sväljningssvårigheter) samt även ökad aktivering av trombocyter och nedsatt fibrinolys. Det sistnämnda ökar risken för tromboser.
, pulmonell hypertension, erektil dysfunktion, spasm i esofagus (sväljningssvårigheter) samt även ökad aktivering av trombocyter och nedsatt fibrinolys. Det sistnämnda ökar risken för tromboser..")
39
Trombocytopeni Trombocyttal under normalområdet (c:a 150 – 400 × 10 9 /L). Symptom – Hudblödningar Petekier < 3 mm Purpura 3–10 mm Ekkymoser > 1 cm – Slemhinneblödningar munhåla, näsa, tarm, urinvägar och uterus – Blödningar i CNS, leder och muskler är ovanligt förekommande.

40
Trombocytopeni Spontana blödningar uppträder oftast först vid TPK < 30 × 10 9 /L Livshotande blödningar ses huvudsakligen vid TPK < 10 × 10 9 /L

41
Orsaker 1.Defekt nybildning i benmärgen Cytostatikabehandling Myelodysplastiskt syndrom Strålning Lymfom/myelom/leukemi Aplastisk anemi B12/folat-brist Alkohol 2.Förstorad mjälte: alla former av splenomegali ger oftast måttlig trombocytopeni 3.Ökad konsumtion och nedbrytning: ITP Sepsis/DIC, TTP

43
Utredning Anemnes – Blödningsanemnes – Andra sjukdomar, infektioner, läkemedel Blodstatus och diff Trombocyter i citrat rör, eller i direktmikroskopi (utesluta pseudotrombocytopeni) CRP, SR, serumelfores HIV och hepatit serologi Benmärgsbiopsi (?) PK, APT tid RF, ANA, antifosfolipidantikroppar Ev. ultraljud mjälte

44
ITP Autoimmun sjukdom – Autoantikroppar mot antigen på trombocytytan (oftast glykoprotein) Labmässigt: Isolerat trombocytopeni av varierande grad Uteslutningsdiagnos: – inga tecken på andra hematologiska sjukdomar, ingen splenomegali, infektion, inflammation, malignitet, alkoholism….. m m

45
Primär och sekundär ITP

46
Behandling - ITP Prednisolon 1-2 mg/kg (bra prognos) Dexametason Immunglobuliner Splenektomi Rituximab Annan immunhämning Trombopoetinanaloger Trombocyttransfusion enbart vid livshotande blödning!
 Dexametason Immunglobuliner Splenektomi Rituximab Annan immunhämning Trombopoetinanaloger Trombocyttransfusion enbart vid livshotande blödning!")
47
Kronisk neutropeni Definitioner Kronisk neutropeni ska ha bestått i minst tre månader för att kallas kronisk. (Ofta övergående neutropeni i samband med viros) Svår neutropeni har absolut blodneutrofilantal <0,5 x 109/L Måttlig neutropeni 0,5-1,0 x 109/L. Lätt neutropeni 1,0-1,5 x 109/L.
 Svår neutropeni har absolut blodneutrofilantal <0,5 x 109/L Måttlig neutropeni 0,5-1,0 x 109/L. Lätt neutropeni 1,0-1,5 x 109/L..")
48
Utredning B12/folat-brist HIV/hepatit Benmärgsprov för att utesluta annan hematologisk sjukdom Flödescytometri på blod, LGL-klon? Mjältförstoring - tex levercirrjos/myeloproliferativ sjukdom. leder till ökad destruktion alkoholanamnes

49
Orsaker Annan autoimmunitet, Felty’s syndrom Autoimmun neutropeni – granulocytantikroppar, att ANC varierar påtagligt, men oregelbundet. Etnisk neutropeni/familjär benign neutropeni LGL Idiopatisk neutropeni Cyklisk neutropeni (prov tas 2 ggr/v under 6 veckors tid) Cyklisk neutropeni kännetecknas av väsentligen normalt ANC, men mycket regelbundet (var 3:e vecka) sjunker ANC till < 0,5 x 10 9 /L under 3-5 dagar. Vid autoimmuna neutropenier är det vanligt Svår kronisk neutropeni ses vid ovanliga medfödda sjukdomar (t ex Kostmanns sjukdom, Shwachman-Diamonds sjukdom
 Cyklisk neutropeni kännetecknas av väsentligen normalt ANC, men mycket regelbundet (var 3:e vecka) sjunker ANC till < 0,5 x 10 9 /L under 3-5 dagar. Vid autoimmuna neutropenier är det vanligt Svår kronisk neutropeni ses vid ovanliga medfödda sjukdomar (t ex Kostmanns sjukdom, Shwachman-Diamonds sjukdom.")
Liknande presentationer






 Förlust av tumörhämmande gener (tumör supressor gener),>")














