Ladda ner presentationen
Presentation laddar. Vänta.

1
Medicinsk utredning Utbildningsdagarna i Lund
Barneurolog Sven Wiklund Barnkliniken i Ystad

2
Disposition Varför Hur Exempel Strategi

3
Varför Prognos; Angelman
Risk för komplikationer; Down, Tuberös skleros Upprepnings risk; Fragile X Förklaring Prognos: Angelman ; Autism + mental retardation AS is caused by disruption of maternally imprinted UBE3A located within the 15q11.2-q13 Angelman syndrome/Prader-Willi syndrome (AS/PWS) region. Risk för komplikationer: Down; VOC tuberös skleros Upprepningsrisk: Fragile X 50% risk för modern att föra CGG repetitionen vidare
 region. Risk för komplikationer: Down; VOC tuberös skleros. Upprepningsrisk: Fragile X 50% risk för modern att föra CGG repetitionen vidare.")
4
Hur Anamnes Somatisk undersökning Radiologi + blodprover ( EEG )
(( EMG + Neurografi ))
)")
5
Anamnes Hereditet Graviditet och Förlossning Tidigare sjukdomar
Nuvarande sjukdom Her:Neurometabol sjd recessiva-kusin gifte, Dystrofia Myotoninca autosom dominant ( förälder ), Fragilt X ( flera i släkten med MR, pojkar svår, flickor lätt ) Grav: infektioner; TORH, missbruk FAS, Förlossning; asyfyxi Tidigare sjukdomar; Meningit; Nuvarande sjukdom: infantil spasm - Aicardi
, Fragilt X ( flera i släkten med MR, pojkar svår, flickor lätt ) Grav: infektioner; TORH, missbruk FAS, Förlossning; asyfyxi. Tidigare sjukdomar; Meningit; Nuvarande sjukdom: infantil spasm - Aicardi.")
6
Förlopp? Utvecklas framåt i sin takt
Stationärt; nått sit utvecklings tak? Fortskridande försämring; Neurometabol sjukdom?Epilepsi? Autism? Hydrocefalus? Hjärntumör? Vaskulär sjukdom/ missbildning? Infektion? Inflammation? Läkemedel? Psykiatri? Failure to Thrive?

7
Hereditet Sju månaders pojke med adducerade tummar bilat sedan födelsen. Fö grav, förlossning samt utveckling normal. Hereditet: Mormor fött fött två barn, som avlidit samt en pojke som har utvecklingstörning. Återbesök vid 10 månader: Fortsatt adducerade tummar, sitter inte utan stöd, hjälper inte vid drag till sittande, inga fallskydds reflexer. Mor nu varit på genetiska kliniken, provtagning för någon sjukdom i släkten, vet ej vilken, hade att göra med adducerade tummar. Sökning PubMed L1 syndromet • Mild to moderate intellectual disability • Delayed onset of speech • Hypotonia progressing to spasticity • Adducted (clasped) thumbs caused by a developmental defect of the extensor pollicis longis and/or brevis muscles • Variable dilatation of the third ventricle X linked L1CAM genen
 thumbs caused by a developmental defect of the extensor pollicis longis and/or brevis muscles. • Variable dilatation of the third ventricle. X linked. L1CAM genen.")
8
Nuvarande sjukdom Aicardis syndrom
Flicka född i ambulans i v 34+6, FV 2680g, Apgar 9,10,10. Intrauterint upptäckt hydrocefalus, MRT visar komplett corpus callosum agenesi, kraftigt vidgadebakhorn samt undergång av vitsubstans runt bakhornen - tidig intrauterin skada? Återbesök 2 månaders ålder ua. remiss till ögonläkare. Ögonläkare undesöker vid 2 1/2 månad. Patologi vid höger papill, blek papill samt rundad näthinneatrofi som tangerar papillkanten, svårbedömt. Återbesök 4 1/2 månad utvecklas jätefint En månad senare Infantil spasm. Aicardis syndrom Sannolikt orsakas syndromet av en mutation på X-kromosomens korta arm, Xp22, men vilket eller vilka arvsanlag (gener) som är förändrade (muterade) är ännu inte känt. Aicardi syndrome appears to be an X-linked dominant disorder with lethality in males, but no gene or candidate region on the X-chromosome has been definitively identified. agenesis of the corpus callosum, distinctive chorioretinal lacunae, and infantile spasms. However, it is now well recognized that several other important findings are typically present in girls with Aicardi syndrome. Neurologic examination can reveal microcephaly, axial hypotonia, and appendicular hypertonia with spasticity. Moderate to severe global developmental delay and intellectual disability are expected. Many girls with Aicardi syndrome develop seizures prior to age three months, and most before age one year. Ongoing medically refractory epilepsy with a variety of seizure types develops over time. Costovertebral defects are common and can lead to marked scoliosis in up to one-third of affected individuals. Other features include characteristic facial features, gastrointestinal difficulties, small hands, vascular malformations and pigmentary lesions of the skin, increased incidence of tumors, lower growth rate after ages seven to nine years, and precocious or delayed puberty. Survival is highly variable, with the mean age of death about 8.3 years and the median age of death about 18.5 years.
 som är förändrade (muterade) är ännu inte känt. Aicardi syndrome appears to be an X-linked dominant disorder with lethality in males, but no gene or candidate region on the X-chromosome has been definitively identified. agenesis of the corpus callosum, distinctive chorioretinal lacunae, and infantile spasms. However, it is now well recognized that several other important findings are typically present in girls with Aicardi syndrome. Neurologic examination can reveal microcephaly, axial hypotonia, and appendicular hypertonia with spasticity. Moderate to severe global developmental delay and intellectual disability are expected. Many girls with Aicardi syndrome develop seizures prior to age three months, and most before age one year. Ongoing medically refractory epilepsy with a variety of seizure types develops over time. Costovertebral defects are common and can lead to marked scoliosis in up to one-third of affected individuals. Other features include characteristic facial features, gastrointestinal difficulties, small hands, vascular malformations and pigmentary lesions of the skin, increased incidence of tumors, lower growth rate after ages seven to nine years, and precocious or delayed puberty. Survival is highly variable, with the mean age of death about 8.3 years and the median age of death about 18.5 years.")
9
Somatisk undersökning
Nijmegen Breakage Syndromesevere microcephaly, with receding forehead and retracted chin and low set ears. Skin abnormalities in the form of café au lait and vitiligo spots are also observed. Most patients are of short stature and underweight (5, 6). A mild to moderate mental retardation, impaired sexual maturation in girls and immunodeficiency are also frequently observed (7). Defective immunological status is characterized by dysgammaglobulinemia with or without decrease in IgG, IgA, IgE, and rarely IgM (8NBS is caused by biallelic mutations in the NBN(formerly called NBS1) gene located on the long arm of chromosome 8. FAS Tuberös Skleros Tuberous sclerosis complex (TSC) involves abnormalities of the skin (hypomelanotic macules, facial angiofibromas, shagreen patches, fibrous facial plaques, ungual fibromas), brain (cortical tubers, subependymal nodules, seizures, intellectual disability), kidney (angiomyolipomas, cysts), and heart (rhabdomyomas, arrhythmias). CNS tumors are the leading cause of morbidity and mortality; renal disease is the second leading cause of early death.Two causative genes, TSC1 and TSC2, have been identified. TSC is inherited in an autosomal dominant manner. Two thirds of affected individuals have TSC as the result of a de novo mutation. CDG manifestations range from severe developmental delay and hypotonia with multiple organ system involvement to hypoglycemia and protein-losing enteropathy with normal development. However, most types have been described in only a few individuals, and thus understanding of the phenotypes is limited. In PMM2-CDG (CDG-Ia), the most common type reported, the clinical presentation and course are highly variable, ranging from death in infancy to mild involvement in adults. Dystrofia Myotonika DM1 is caused by expansion of a CTG trinucleotide repeat in the non-coding region of DMPK.multisystem disorder that affects skeletal and smooth muscle as well as the eye, heart, endocrine system, and central nervous system. The clinical findings, which span a continuum from mild to severe, have been categorized into three somewhat overlapping phenotypes: mild, classic, and congenital. Mild DM1 is characterized by cataract and mild myotonia (sustained muscle contraction); life span is normal. Classic DM1 is characterized by muscle weakness and wasting, myotonia, cataract, and often cardiac conduction abnormalities; adults may become physically disabled and may have a shortened life span. Congenital DM1 is characterized by hypotonia and severe generalized weakness at birth, often with respiratory insufficiency and early death; intellectual disability is common. Nijmegen Breakage Syndrome FAS Tuberös Skleros CDG Dystrofia Myotonica
. A mild to moderate mental retardation, impaired sexual maturation in girls and immunodeficiency are also frequently observed (7). Defective immunological status is characterized by dysgammaglobulinemia with or without decrease in IgG, IgA, IgE, and rarely IgM (8NBS is caused by biallelic mutations in the NBN(formerly called NBS1) gene located on the long arm of chromosome 8. FAS. Tuberös Skleros Tuberous sclerosis complex (TSC) involves abnormalities of the skin (hypomelanotic macules, facial angiofibromas, shagreen patches, fibrous facial plaques, ungual fibromas), brain (cortical tubers, subependymal nodules, seizures, intellectual disability), kidney (angiomyolipomas, cysts), and heart (rhabdomyomas, arrhythmias). CNS tumors are the leading cause of morbidity and mortality; renal disease is the second leading cause of early death.Two causative genes, TSC1 and TSC2, have been identified. TSC is inherited in an autosomal dominant manner. Two thirds of affected individuals have TSC as the result of a de novo mutation. CDG manifestations range from severe developmental delay and hypotonia with multiple organ system involvement to hypoglycemia and protein-losing enteropathy with normal development. However, most types have been described in only a few individuals, and thus understanding of the phenotypes is limited. In PMM2-CDG (CDG-Ia), the most common type reported, the clinical presentation and course are highly variable, ranging from death in infancy to mild involvement in adults. Dystrofia Myotonika DM1 is caused by expansion of a CTG trinucleotide repeat in the non-coding region of DMPK.multisystem disorder that affects skeletal and smooth muscle as well as the eye, heart, endocrine system, and central nervous system. The clinical findings, which span a continuum from mild to severe, have been categorized into three somewhat overlapping phenotypes: mild, classic, and congenital. Mild DM1 is characterized by cataract and mild myotonia (sustained muscle contraction); life span is normal. Classic DM1 is characterized by muscle weakness and wasting, myotonia, cataract, and often cardiac conduction abnormalities; adults may become physically disabled and may have a shortened life span. Congenital DM1 is characterized by hypotonia and severe generalized weakness at birth, often with respiratory insufficiency and early death; intellectual disability is common. Nijmegen Breakage Syndrome. FAS. Tuberös Skleros. CDG. Dystrofia Myotonica.")
10
Somatisk Undersökning
Supra valvulär aorta stenos - Williams syndrom deletion) av den långa armen på en av de två kromosomerna i kromosompar 7 (7q11.23). Oftast nymutaion,Ärftlighetsgången är då autosomal dominant,Williams syndrome (WS) is characterized by cardiovascular disease (elastin arteriopathy, peripheral pulmonary stenosis, supravalvular aortic stenosis, hypertension), distinctive facies, connective tissue abnormalities, intellectual disability (usually mild), a specific cognitive profile, unique personality characteristics, growth abnormalities, and endocrine abnormalities (hypercalcemia, hypercalciuria, hypothyroidism, and early puberty). Feeding difficulties often lead to failure to thrive in infancy. Hypotonia and hyperextensible joints can result in delayed attainment of motor milestones. Supravalvulär Aorta Stenos Williams Syndrom
 av den långa armen på en av de två kromosomerna i kromosompar 7 (7q11.23). Oftast nymutaion,Ärftlighetsgången är då autosomal dominant,Williams syndrome (WS) is characterized by cardiovascular disease (elastin arteriopathy, peripheral pulmonary stenosis, supravalvular aortic stenosis, hypertension), distinctive facies, connective tissue abnormalities, intellectual disability (usually mild), a specific cognitive profile, unique personality characteristics, growth abnormalities, and endocrine abnormalities (hypercalcemia, hypercalciuria, hypothyroidism, and early puberty). Feeding difficulties often lead to failure to thrive in infancy. Hypotonia and hyperextensible joints can result in delayed attainment of motor milestones. Supravalvulär Aorta Stenos Williams Syndrom.")
11
Somatisk Undersökning
Sjukdomen orsakas av brist på enzymet glukosylceramidas (andra benämningar är glukocerebrosidas, cerebrosid-ß-glukosidas, E.C ). Enzymbristen gör att nedbrytningen av glukosylceramid inte fungerar, utan detta fettliknande ämne inlagras, framför allt i en speciell typ av vita blodkroppar (makrofager). Makrofager fyllda med glukosylceramid kallas Gaucherceller och ansamlas i olika organ i kroppen. Arvsanlaget (genen) för enzymet sitter på långa armen på kromosom 1 och betecknas GBA. Mer än 300 skador (mutationer) i genen som leder till Gauchers sjukdom har påvisats. Splenomegali Gauchers sjukdom
. Enzymbristen gör att nedbrytningen av glukosylceramid inte fungerar, utan detta fettliknande ämne inlagras, framför allt i en speciell typ av vita blodkroppar (makrofager). Makrofager fyllda med glukosylceramid kallas Gaucherceller och ansamlas i olika organ i kroppen. Arvsanlaget (genen) för enzymet sitter på långa armen på kromosom 1 och betecknas GBA. Mer än 300 skador (mutationer) i genen som leder till Gauchers sjukdom har påvisats. Splenomegali Gauchers sjukdom.")
12
Somatisk Undersökning
Noonan syndrome (NS) is characterized by short stature, congenital heart defect, and developmental delay of variable degree. Other findings can include broad or webbed neck, unusual chest shape with superior pectus carinatum and inferior pectus excavatum, cryptorchidism, characteristic facies, varied coagulation defects, lymphatic dysplasias, and ocular abnormalities. Although birth length is usually normal, final adult height approaches the lower limit of normal. Congenital heart disease occurs in 50%-80% of individuals. Pulmonary valve stenosis, often with dysplasia, is the most common heart defect and is found in 20%-50% of individuals. Hypertrophic cardiomyopathy, found in 20%-30% of individuals, may be present at birth or develop in infancy or childhood. Other structural defects include atrial and ventricular septal defects, branch pulmonary artery stenosis, and tetralogy of Fallot. Up to one third of affected individuals have mild intellectual disability. Affected individuals have normalchromosome studies. Molecular genetic testing identifies a mutation in PTPN11 in 50% of affected individuals, SOS1 in approximately 13%, RAF1 in 3% to 17%, and KRAS in fewer than 5%. Other genes in which mutations have been reported Genetic counseling. NS is inherited in an autosomal dominant manner. Although many individuals with NS have a de novomutation, an affected parent is recognized in 30%-75% of families. Retentio Testis Noonans Syndrom
 is characterized by short stature, congenital heart defect, and developmental delay of variable degree. Other findings can include broad or webbed neck, unusual chest shape with superior pectus carinatum and inferior pectus excavatum, cryptorchidism, characteristic facies, varied coagulation defects, lymphatic dysplasias, and ocular abnormalities. Although birth length is usually normal, final adult height approaches the lower limit of normal. Congenital heart disease occurs in 50%-80% of individuals. Pulmonary valve stenosis, often with dysplasia, is the most common heart defect and is found in 20%-50% of individuals. Hypertrophic cardiomyopathy, found in 20%-30% of individuals, may be present at birth or develop in infancy or childhood. Other structural defects include atrial and ventricular septal defects, branch pulmonary artery stenosis, and tetralogy of Fallot. Up to one third of affected individuals have mild intellectual disability. Affected individuals have normalchromosome studies. Molecular genetic testing identifies a mutation in PTPN11 in 50% of affected individuals, SOS1 in approximately 13%, RAF1 in 3% to 17%, and KRAS in fewer than 5%. Other genes in which mutations have been reported Genetic counseling. NS is inherited in an autosomal dominant manner. Although many individuals with NS have a de novomutation, an affected parent is recognized in 30%-75% of families. Retentio Testis Noonans Syndrom.")
13
Somatisk Undersökning
Woods Light Tuberös Skleros

14
Blodprover PKU: Arginasbrist, Isovaleriansyrauri, Argininosuccinatlyasbrist,LCHAD-Brist, Betaketotiolasbrist, MSUD, Biotinidasbrist, MCAD-brist, CACT, Medfödd binjurebarkshyperplasi, Citrullinemi, Medfödd hypothyreos, CPT1 och 2, Metylmalonsyruri, fenylketonuri, Primär Karnitinbrist, Galaktosemi, Propionsyruri, Glutarsyrauri typ 1 och 2, Tyrosinemi typ 1, VLCAD-brist, Homocystinuri Blodstatus: Fanconis anemi; Mukolipidos Typ 4 TSH/ T4 fritt: Downs Syndrom S-laktat; Mitokondriella sjukdomar, Leighs sjukdom S-Ca; Williams Syndrom, 22q11 Deletions Syndromet CDT; CongenitalDefectofGlycosylation CK; Dystrofia myotonica Fanconi: låga röda, vita, trcc. Mukolipidos typ 4 järnbrist anemi CK normalt eller lätt förhöjt vid DM Hypocalcemi vid William samt CATCH 22 S Laktat mitokondriella sjukdomar CDG-syndromet beror på en störning i glykosyleringen (bildningen av sammansatta sockerkedjor) i vissa glykoproteiner (kemiska föreningar bestående av protein och kolhydrat) med viktiga biologiska funktioner.törningen i ämnesomsättningen leder till skador i lillhjärnan och hjärnstammen (olivopontocerebellär atrofi), perifera nerverna, näthinnan, skelettet, levern och könskörtlarna. Även hjärtat, njurarna och bukspottkörteln kan påverkas. In the setting of existing newborn screening programs for congenital hypothyroidism, screening of children with developmental delay with thyroid function studies is not indicated unless
 i vissa glykoproteiner (kemiska föreningar bestående av protein och kolhydrat) med viktiga biologiska funktioner.törningen i ämnesomsättningen leder till skador i lillhjärnan och hjärnstammen (olivopontocerebellär atrofi), perifera nerverna, näthinnan, skelettet, levern och könskörtlarna. Även hjärtat, njurarna och bukspottkörteln kan påverkas. In the setting of existing newborn screening programs. for congenital hypothyroidism, screening of. children with developmental delay with thyroid. function studies is not indicated unless.")
15
Blodprover Spinal- laktat, Glukos, protein, ev skademärkörer
Elfores; NejmigenBreakage Syndrome Homocystein, B-12 ( Borrelia Serologi ) Gluten serologi Inborn Error of metabolism Genetik Spinal- laktat, Glukos, protein, ev skademärkörer NBS: • Agammaglobulinemia has been found in 35% and IgA deficiency in 20% of affected individuals. • Deficiencies in IgG2 and IgG4 are frequent even when the IgG serum concentration is normal. • The most commonly reported defects in cellular immunity are reduced percentages of total CD3+ T cells and CD4+ cells. • An increased frequency of T cells with a memory phenotype (CD45RO+) and a concomitant decrease in naive T cells (CD45RA+) has been reported [Michalkiewicz et al Given the low yield of about 1%, routine metabolic screening for inborn errors of metabolism2003]. Metabolic testing may be pursued in the context of historical (parental consanguinity, family history, developmental regression, episodic decompensation) or physical examination findings that are suggestive of a specific etiology Routine cytogenetic testing (yield of 3.7%) is indicated in the evaluation of the child with developmental delay even in the absence of dysmorphic features or clinical features Testing for the fragile X mutation (yield of 2.6%), particularly in the presence of a family history of developmental delay, may be considered in the evaluation of the child with global developmental delay. The diagnosis of Rett syndrome should be considered in females with unexplained moderate to severe mental retardation. People with GLUT1 deficiency syndrome may have developmental delay or intellectual disability. They may also have other neurological problems, such as stiffness caused by abnormal tensing of the muscles (spasticity), difficulty in coordinating movements (ataxia), and speech difficulties (dysarthria). Some experience episodes of confusion, lack of energy (lethargy), headaches, muscle twitches (myoclonus), or involuntary irregular eye movements, particularly before meals.The SLC2A1 gene provides instructions for producing a protein called the glucose transporter protein type 1 (GLUT1). This protein is part of the membranes of cells, where it transports glucose (a simple sugar) from the blood into the cells for use as fuel. Glucose transporter protein type 1 is involved in moving glucose across the blood- brain barrier,
 Gluten serologi. Inborn Error of metabolism. Genetik. Spinal- laktat, Glukos, protein, ev skademärkörer. NBS: • Agammaglobulinemia has been found in 35% and IgA deficiency in 20% of affected individuals. • Deficiencies in IgG2 and IgG4 are frequent even when the IgG serum concentration is normal. • The most commonly reported defects in cellular immunity are reduced percentages of total CD3+ T cells and CD4+ cells. • An increased frequency of T cells with a memory phenotype (CD45RO+) and a concomitant decrease in naive T cells (CD45RA+) has been reported [Michalkiewicz et al Given the low yield of about 1%, routine metabolic. screening for inborn errors of metabolism2003]. Metabolic testing. may be pursued in the context of historical (parental. consanguinity, family history, developmental. regression, episodic decompensation) or. physical examination findings that are suggestive. of a specific etiology. Routine cytogenetic testing (yield of 3.7%) is indicated. in the evaluation of the child with developmental. delay even in the absence of dysmorphic features or clinical features. Testing for the fragile X mutation (yield of 2.6%), particularly in the presence of a family history of. developmental delay, may be considered in the. evaluation of the child with global developmental. delay. The diagnosis of Rett syndrome should be considered. in females with unexplained moderate to severe. mental retardation. People with GLUT1 deficiency syndrome may have developmental delay or intellectual disability. They may also have other neurological problems, such as stiffness caused by abnormal tensing of the muscles (spasticity), difficulty in coordinating movements (ataxia), and speech difficulties (dysarthria). Some experience episodes of confusion, lack of energy (lethargy), headaches, muscle twitches (myoclonus), or involuntary irregular eye movements, particularly before meals.The SLC2A1 gene provides instructions for producing a protein called the glucose transporter protein type 1 (GLUT1). This protein is part of the membranes of cells, where it transports glucose (a simple sugar) from the blood into the cells for use as fuel. Glucose transporter protein type 1 is involved in moving glucose across the blood- brain barrier,")
16
Öga Kolobom; CHARGE Cherry Red Spot; Tay Sachs
A cherry-red spot is a finding in the macula of the eye in a variety of lipid storage disorders and in central retinal artery occlusion.[1] It describes the appearance of a small circular choroid shape as seen through the fovea centralis. [2] Its appearance is due to a relative transparency of the macula; storage disorders cause the accumulation of storage material within the cell layers of the retina, however, the macula, which is relatively devoid of cellular layers, does not build up this material, and thus allows the eye to see through the macula to the red choroid below. [3] Lysosomal sjukdomGM2 gangliosidosis or Hexosaminidase A deficiency) is an autosomal recessivegenetic disorder. In its most common variant, known as infantile Tay–Sachs disease, it causes a relentless deterioration of mental and physical abilities that commences around six months of age and usually results in death by the age of four.[1] Because Aicardi syndrome is seen only in females and 47,XXY males, it is presumed to be caused by adominant de novo mutation in an X-linked gene with lethality in 46,XY males.Aicardi syndrome was classically characterized by a triad of features: agenesis of the corpus callosum, distinctive chorioretinal lacunae, and infantile spasms. However, it is now well recognized that several other important findings are typically present in girls with Aicardi syndrome. Neurologic examination can reveal microcephaly, axial hypotonia, and appendicular hypertonia with spasticity. Moderate to severe global developmental delay and intellectual disability are expected. CHARGE is a mnemonic that stands for coloboma, heart defects, choanal atresia, retarded growth and development, genital abnormalities, and ear anomalies. CHARGE syndrome is characterized by unilateral or bilateral coloboma of the iris, retina-choroid, and/or disc with or without microphthalmos (80%-90% of individuals); unilateral or bilateral choanal atresia or stenosis (50%-60%); cranial nerve dysfunction resulting in hyposmia or anosmia, unilateral or bilateral facial palsy (40%), impaired hearing and/or swallowing problems (70%-90%); abnormal outer ears, ossicular malformations, Mondini defect of the cochlea and absent or hypoplastic semicircular canals (>90%); cryptorchidism in males and hypogonadotrophic hypogonadism in both males and females; developmental delay; cardiovascular malformations (75%-85%); growth deficiency (70%-80%); orofacial clefts (15%-20%); and tracheoesophageal fistula (15%-20%). Neonates with CHARGE syndrome often have multiple life-threatening medical conditions. Feeding difficulties are a major cause of morbidity in all age groups.CHD7, encoding the chromodomain helicase DNA binding protein, is the only gene currently known to be associated with CHARGE syndrome.Sequence analysis of the CHD7 coding region detects mutations in approximately 60%-70% of individuals with CHARGE syndrome. Cherry Red Spot; Tay Sachs Chorioretinala Lakuner; Aicardi Kolobom; CHARGE
 is an autosomal recessivegenetic disorder. In its most common variant, known as infantile Tay–Sachs disease, it causes a relentless deterioration of mental and physical abilities that commences around six months of age and usually results in death by the age of four.[1] Because Aicardi syndrome is seen only in females and 47,XXY males, it is presumed to be caused by adominant de novo mutation in an X-linked gene with lethality in 46,XY males.Aicardi syndrome was classically characterized by a triad of features: agenesis of the corpus callosum, distinctive chorioretinal lacunae, and infantile spasms. However, it is now well recognized that several other important findings are typically present in girls with Aicardi syndrome. Neurologic examination can reveal microcephaly, axial hypotonia, and appendicular hypertonia with spasticity. Moderate to severe global developmental delay and intellectual disability are expected. CHARGE is a mnemonic that stands for coloboma, heart defects, choanal atresia, retarded growth and development, genital abnormalities, and ear anomalies. CHARGE syndrome is characterized by unilateral or bilateral coloboma of the iris, retina-choroid, and/or disc with or without microphthalmos (80%-90% of individuals); unilateral or bilateral choanal atresia or stenosis (50%-60%); cranial nerve dysfunction resulting in hyposmia or anosmia, unilateral or bilateral facial palsy (40%), impaired hearing and/or swallowing problems (70%-90%); abnormal outer ears, ossicular malformations, Mondini defect of the cochlea and absent or hypoplastic semicircular canals (>90%); cryptorchidism in males and hypogonadotrophic hypogonadism in both males and females; developmental delay; cardiovascular malformations (75%-85%); growth deficiency (70%-80%); orofacial clefts (15%-20%); and tracheoesophageal fistula (15%-20%). Neonates with CHARGE syndrome often have multiple life-threatening medical conditions. Feeding difficulties are a major cause of morbidity in all age groups.CHD7, encoding the chromodomain helicase DNA binding protein, is the only gene currently known to be associated with CHARGE syndrome.Sequence analysis of the CHD7 coding region detects mutations in approximately 60%-70% of individuals with CHARGE syndrome. Cherry Red Spot; Tay Sachs. Chorioretinala Lakuner; Aicardi. Kolobom; CHARGE.")
17
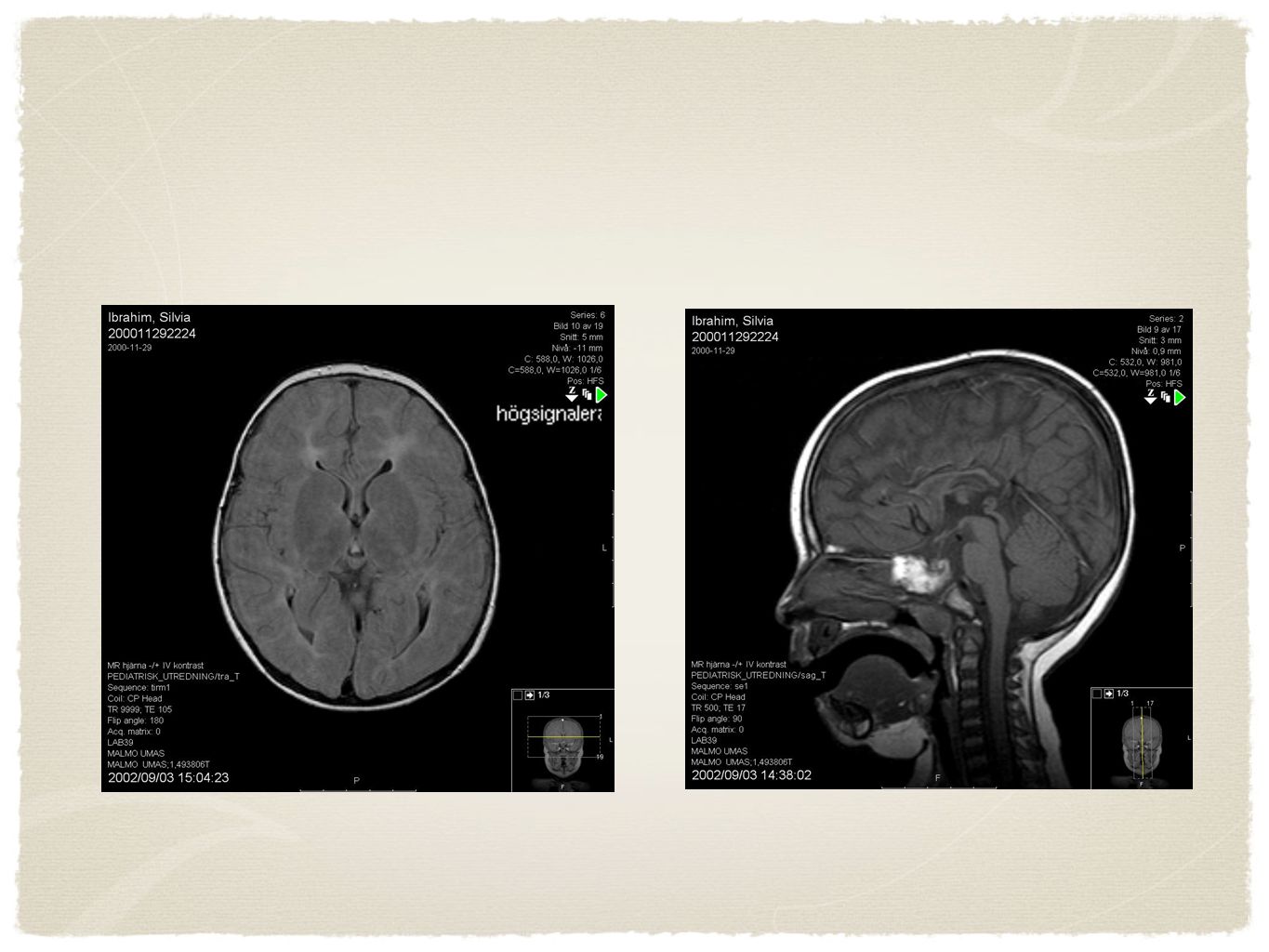
Radiologi Två årig flicka, utvecklingsmässigt på ett årings nivå. Epikantusveck. Ingen fortskridande försämring MRT visar hypoplastisk corpus callosum, supratentoriell reducering av vita substansen med patologisk förhöjd T2 signal, Bild som vid Leukodystrofi, t.ex. mukolipidos typ 4. Förflyttar sig genom att hasa på magen, svårt att grappa föremål, sväljnings svårigheter, inget tal eller tecken användning men interagerar aktivt. Har epilepsi. Neuroimaging is recommended as part of the diagnostic evaluation of the child with global developmental delay (Level B; class III evidence). As the presence of physical findings (e.g., microcephaly, focal motor findings) increases the yield of making a specific neuroimaging diagnosis, physicians can more readily consider obtaining a scan in this population
. As. the presence of physical findings (e.g., microcephaly, focal motor findings) increases the yield of. making a specific neuroimaging diagnosis, physicians. can more readily consider obtaining a scan. in this population.")
19
Radiologi Prospektiv studie, 157 barn.Major/Specific findings Brain Injury associatedwith prematurity12 Periventricular Leukomalacia (12) Brain malformations 7 Abnormal Ventral Induction Semilobar Holoprosencephaly (1) Abnormal Commissuration Hypoplasia of Corpus callosumwith lipoma (1) Abnormal Cortical formation Heterotopia (1) Focal cortical dysplasia (2) Schizencephaly and septo-opticdysplasia (1) Posterior Fossa Malformation Chiari 1 (1) Focal gliosis of unknown cause 4 Previous infection 2 Toxic/Metabolic 2 Previous hypoglycaemicinjury (2) Hydrocephalus 1 Phakomatoses 1 Tuberous sclerosis complex (1) Total 29 This prospective study was performed as part of an audit study registered at Sheffield Children’s Hospital, Sheffield, England under the auspices of investigating improvements in patient care. The specific aspect of this study was to look at the role of a detailed MR imaging protocol and MR spectroscopy for children being investigated for DD. All children referred for MR imaging because of developmental delay were included in the study over an 18-month period commencing in January The children came from three sources; from paediatric and paediatric neurology departments within a dedicated children’s hospital, from paediatric departments from district general hospitals and from community paediatricians. MR protocol We performed MR imaging and spectroscopy protocol on 157 children with developmental delay.
 Brain malformations 7 Abnormal Ventral Induction Semilobar Holoprosencephaly (1) Abnormal Commissuration Hypoplasia of Corpus callosumwith lipoma (1) Abnormal Cortical formation Heterotopia (1) Focal cortical dysplasia (2) Schizencephaly and septo-opticdysplasia (1) Posterior Fossa Malformation Chiari 1 (1) Focal gliosis of unknown cause 4 Previous infection 2 Toxic/Metabolic 2 Previous hypoglycaemicinjury (2) Hydrocephalus 1 Phakomatoses 1 Tuberous sclerosis complex (1) Total 29. This prospective study was performed as part of an audit. study registered at Sheffield Children’s Hospital, Sheffield, England under the auspices of investigating improvements. in patient care. The specific aspect of this study was to look. at the role of a detailed MR imaging protocol and MR. spectroscopy for children being investigated for DD. All. children referred for MR imaging because of developmental. delay were included in the study over an 18-month period. commencing in January The children came from. three sources; from paediatric and paediatric neurology. departments within a dedicated children’s hospital, from. paediatric departments from district general hospitals and. from community paediatricians. MR protocol. We performed MR imaging and. spectroscopy protocol on 157 children with developmental. delay.")
20
EEG AICARDI ESES Absens epilepsi
Epileptic status eplilepticus during slow sleep An EEG can be obtained when a child with global developmental delay has a history or examination features suggesting the presence of epilepsy or a Data are insufficient to permit making a recommendation regarding the role of EEG in a child with global developmental delay in whom there is no clinical evidence of epilepsy

21
Strategi Noggran anamnes Noggran undersökning
Dagis och Skolbarn, friskt, inga stigmata, ingen hereditet, inget i status - Ingen utredning Tre minor signs eller fler, hereditet; Genetisk utredning Avvikande Neurologi; Överväg MRT Spädbarn; Om progressivt “Rubbet” och det snabbt Spädbarn; Sen men utvecklas framåt utan något uppenbart i anamnes eller status; aktiv expektans, utred stegvis och intelligent.

Liknande presentationer